
Lupus eritematoso sistémico
| Lupus eritematoso sistémico | ||
|---|---|---|
 | ||
| Especialidad | Reumatología | |
| Síntomas | Dolor e inflamación articular, fiebre, dolor torácico, pérdida del cabello, úlceras bucales, inflamación de los ganglios linfáticos, fatiga, eflorescencia[1][2] | |
| Inicio habitual | 15—45 años de edad[1][3] | |
| Duración | Crónica[2] | |
| Causas | Desconocidas[1] | |
| Factores de riesgo | Sexo femenino[2] | |
| Diagnóstico | Basado en los síntomas y en análisis de sangre[2][1] | |
| Medicación | AINEs, corticoesteroides, inmunosupresores, hidroxicloroquina, metotrexato[1][2] | |
| Pronóstico | Supervivencia del 85-90% 10 años tras el comienzo de la enfermedad[2] | |
| Frecuencia | Incidencia de 5,6 por cada 100 000 habitantes[2] | |
| Sinónimos | ||
| ||
El lupus eritematoso sistémico (LES) es una de las enfermedades autoinmunitarias más prevalentes; es crónica, caracterizada por un curso cíclico donde se alternan períodos de exacerbaciones y remisiones; sistémica, ya que afecta prácticamente cualquier órgano del cuerpo; y heterogénea, puesto que el espectro de manifestaciones clínicas y alteraciones serológicas es muy amplio y variado. La mayoría de los casos presenta un cuadro clínico leve o moderado; sin embargo, puede presentarse o desarrollarse con un compromiso grave de órganos vitales.[4][5][6]
El LES es la enfermedad autoinmunitaria no organoespecífica por excelencia, ya que el organismo produce numerosos autoanticuerpos dirigidos contra antígenos celulares, cuyo resultado final son lesiones inflamatorias de múltiples órganos y sistemas. Principalmente, se afectan los riñones, la piel y las mucosas, el sistema músculo esquelético, el sistema cardiovascular, el sistema nervioso y el sistema respiratorio.[6][7][8]
Su etiología se desconoce; sin embargo, varios estudios señalan ciertas alteraciones genéticas predisponentes que desencadenan la enfermedad frente a estímulos ambientales aún no precisados.[7]
El LES es una enfermedad crónica, con un curso clínico heterogéneo, variando desde formas clínicas relativamente benignas hasta cuadros graves con riesgo para la vida. Los objetivos del tratamiento son asegurar la supervivencia a largo plazo, mantener la actividad de la enfermedad lo más bajo posible, prevenir el daño orgánico, minimizar los efectos adversos del tratamiento, mejorar la calidad de vida e informar a los pacientes acerca de su rol en mantener la enfermedad bajo control. Los pacientes requieren ser controlados por un reumatólogo en forma regular, y en caso de tener compromiso de diversos órganos, de todo un equipo multidisciplinario.[9]
El 10 de mayo se celebra el Día Mundial de la lucha contra el Lupus.[10]
Etimología
[editar]El origen del nombre de la enfermedad no es del todo conocido. El término lupus (lobo, en latín) fue el nombre de una familia romana y existió un santo Lupus que vivió en Francia en el año 600.[11]
Se considera que la primera aparición del término lupus en relación con la salud se debe a Herbemius de Tours, a finales del siglo X, que describe la curación de Hildricus, obispo de Lieja, en el santuario de San Martín de Tours: «[...] gravemente afectado por la enfermedad denominada lupus».[12]
Entre los siglos XV y XVIII, las lesiones cutáneas faciales y otras enfermedades destructivas, como la tuberculosis cutánea, se denominaron lupus por la similitud con la mordedura de este animal.[11][13][14]
El término eritematoso (del griego: ερυθρός, erythros) significa rojo o enrojecido. Se atribuye a Cazenave en 1851 la introducción de la denominación lupus eritematoso; referido al enrojecimiento o eritema malar en forma de alas de mariposa.[15]
El término sistémico se refiere a la afectación difusa o generalizada de la enfermedad —no solo cutánea—, con alteraciones en múltiples órganos o sistemas. Fue William Oster quien, entre 1872 y 1895, se refirió por primera vez a esta enfermedad como diseminada o sistémica.[11]
En 1954, Harvey suprimió el término "diseminado", y quedó «lupus eritematoso sistémico» como denominación fundamental de la enfermedad a partir de entonces.[16]
Historia
[editar]
Es probable que algunas descripciones de lesiones cutáneas realizadas por Hipócrates en el siglo V a. C. se correspondieran con lesiones lúpicas. Las llamó herpes esthiomenos que significa dermatosis persistente. El uso más antiguo del término lupus en su acepción médica se sitúa entre los años 916 y 963 y fue realizado por Herbemius de Tours, refiriéndose a lesiones cutáneas.[12][13][15]
Rogerius Frugardi, en 1230,usó el término lupus para describir lesiones faciales erosivas, y Giovanni Manardi, en 1530, usó el mismo término para indicar forúnculos y ulceraciones de las extremidades inferiores.[13]
Rudolph Virchow, patólogo alemán, después de revisar la literatura de la Edad Media y el Renacimiento, concluyó que cualquier proceso que involucrara ulceración o necrosis de las extremidades inferiores era denominado lupus independientemente de la causa que las producía. En este cajón se encontraban además de las lesiones cutáneas propiamente lúpicas, la tuberculosis, la sífilis o el cáncer cutáneos.[13]
Robert Willan (1757-1812), dermatólogo británico, describió lesiones destructivas de la cara y nariz bajo la denominación de lupus. Se incluían aquí la tuberculosis cutánea o lupus vulgar. Separó así este tipo de lesiones de las vesiculares que denominó herpes. Thomas Bateman, uno de sus estudiantes, completó su trabajo y agrupó las lesiones destructivas en la denominación de lupus willani.[12][13]
La primera descripción clara del lupus eritematoso se atribuye a Laurent Theodore Biett, de la escuela parisina de dermatología, que la llamó eritema centrifugum. Entre 1833 y 1851, su estudiante Pierre Louis Alphee Cazenave (1795-1877) publicó su trabajo y acuñó el término lupus eritematoso, describiéndolo como una condición rara que se presentaba en mujeres jóvenes previamente sanas, y que afectaba principalmente la cara. Las describe como lesiones enrojecidas levemente solevantadas del tamaño de una moneda que crecían gradualmente hasta extenderse en gran parte de la cara; los bordes eran prominentes y su centro iba retornando a su color normal, sin dolor ni picor. Probablemente Cazenave describía el lupus discoide, después llamado "lupus eritematoso cutáneo".[12][13][17]
Ferdinand von Hebra describió, entre 1845 y 1866, la erupción que aparecía, en forma de alas de mariposa, como una erupción «[...] principalmente en la cara, las mejillas y la nariz, con una distribución similar a una mariposa». Inicialmente denominó este padecimiento seborrea congestiva.[12][13][18]
A finales del siglo XIX (1872), Moritz Kaposi describió por primera vez algunos signos sistémicos de la enfermedad (pérdida de peso, fiebre, anemia, linfadenopatía y artritis) y distinguió la forma exclusivamente cutánea de la enfermedad, a la que denominó lupus discoide, hoy denominado lupus eritematoso cutáneo. Kaposi, al igual que Cazenave, diferencia esta enfermedad de la tuberculosis cutánea (lupus vulgar).[12][13][19]
Posteriormente William Osler, entre 1895 y 1903, describió ya la mayoría de las complicaciones viscerales del lupus, acuñando el término lupus eritematoso sistémico, y el lupus dejó de ser una enfermedad cutánea para pasar a ser una enfermedad con afectación de múltiples órganos.[13][15][18]

En 1894, Payne, del hospital St. Thomas de Londres, divulgó la utilidad de la quinina en el tratamiento del lupus y señaló una causa vascular como subyacente a la enfermedad. Cuatro años más tarde, el uso de salicilatos en asociación con quinina demostró ser todavía más beneficioso.[12][13]
En 1902, Jonathan Hutchinson describió la naturaleza fotosensitiva de las lesiones cutáneas faciales. Sequira y Balean describieron la acroasfixia o fenómeno de Raynaud, y la nefritis lúpica.[13]
En 1904, Jadassohn realizó un estudio exhaustivo del lupus discoide y sistémico, contribuyendo grandemente al entendimiento de la enfermedad.[12][13]
En 1908, Alfred Kraus y Carl Bohac describieron el compromiso pulmonar del lupus.[13]
En 1923, Emanuel Libman y Benjamin Sacks describieron la endocarditis asociada al lupus. George Belote y H. S. Ratner confirmaron que la endocarditis de Libman-Sacks era una manifestación del lupus sin compromiso cutáneo, rompiendo la idea de que el lupus siempre se presentaba con lesiones en la piel.[13]
En 1935, Paul Klemperer, George Baehr y A. D. Pollack describieron la lesión típica en «asa de alambre» presente en la nefritis lúpica.[13]
En 1948, Malcolm Hargraves descubrió las células LE (de "lupus eritematoso"), lo que sirvió para establecer con ciertas garantías el diagnóstico de la enfermedad. Posteriormente, se demostró que este factor era un anticuerpo antinuclear.[15][18]
A mediados del siglo XX, los trabajos de Philip Showalter Hench de la Clínica Mayo sobre la eficacia de los corticoides en las enfermedades reumáticas revolucionaron el tratamiento del lupus. Por ello, se le concedió el premio Nobel de Fisiología y Medicina en 1950.[13]
En 1954, Harvey acuñó el nombre lupus eritematoso sistémico.[cita requerida]
En 1971 la Asociación Estadounidense del Reumatismo (ARA) publicó los primeros criterios de clasificación del LES, que se revisaron posteriormente en 1982, 1997 y 2012.[15]
Epidemiología
[editar]La prevalencia de LES en la población general es de 20 a 150 casos por cada 100 000 habitantes —dependiendo de la zona geográfica y el origen étnico—.[20] Factores dependientes del sexo y la raza influyen de modo notable en la mayor o menor incidencia de la enfermedad en diversos subgrupos. Así, la prevalencia en mujeres caucásicas es de 164 por 100 000, frente a 406 por 100 000 en las mujeres afroamericanas.[21]
- Sexo: la incidencia del LES es 10 veces más alta en las mujeres que en los hombres,[19] correspondiendo el 90 % de los casos a mujeres en edad reproductiva.[21] La mayor prevalencia en mujeres se atribuye, al menos en parte, al efecto hormonal de los estrógenos; así en niños —en los que el efecto hormonal es presumiblemente pequeño— la relación mujer/hombre es 3:1, en los adultos —en especial en las mujeres en edad de procrear— es de 7:1 a 15:1, y en los individuos de más edad —en especial mujeres postmenopáusicas— es de 8:1.[22]
- Factores étnicos: el LES tiene una distribución universal, afectando a todas las razas y estando presente en todos los continentes. Sin embargo, de igual modo que hay diferencias según el sexo, también hay diferencias raciales y geográficas. Así, el LES es 2 a 3 veces más frecuente en las mujeres afrodescendientes que en las caucásicas,[23] aunque, en comparación, el LES ocurre con menor frecuencia en los negros en África que en Norte América.[24]
- Edad de inicio: el comienzo de la enfermedad en el 65 % de los casos es entre los 16 y 65 años, frente a un 20 % de inicio antes de los 16 años y un 15 % después de los 65 años. Las manifestaciones clínicas de la enfermedad y la gravedad de las mismas pueden ser distintas en diversos subgrupos según la raza, el sexo y la edad de comienzo.[21] Así, las manifestaciones clínicas del lupus tienden a ser más leves en los pacientes de más edad, con una forma de presentación similar a la observada en el lupus eritematoso inducido por fármacos.[25]
Etiología
[editar]La causa exacta del lupus eritematoso sistémico es desconocida, aunque parece claramente multifactorial. Diversas observaciones sugieren la intervención de factores genéticos, hormonales y ambientales, que pueden influir en el sistema inmunitario y provocar el cuadro clínico del LES. En los pacientes con lupus hay numerosos defectos inmunes. Sin embargo, la etiología de estas anomalías no está clara, no conociéndose qué defectos son primarios y cuáles son inducidos secundariamente.[21]
Factores genéticos
[editar]
La observación de una elevada concordancia en la aparición de LES en gemelos monocigotos —entre el 15 y 57 %— y una mayor prevalencia de la enfermedad —del 5 al 12 %— entre descendientes de pacientes con LES, son compatibles con el importante papel de la genética en la patogénesis del lupus.[21] El LES es una enfermedad multigénica, no existiendo un polimorfismo de un único gen que origine un elevado riesgo de la enfermedad, excepto para la rara mutación TREX1 o las deficiencias de los componentes tempranos del sistema del complemento —C1q, C2 y C4—.[21]
Los genes más habituales que predisponen al lupus se ubican en la región del HLA (antígenos leucocitarios humanos), especialmente en genes con HLA de clase II DR y DQ, incluyendo HLA-DR2 y HLA-DR3, así como en genes de clase III que codifican algunos componentes del sistema del complemento. Algunas proteínas que son importantes para eliminar las células apoptóticas también participan en la predisposición genética; por ejemplo las deficiencias homocigotas de los primeros componentes del complemento C1q, C2 y C4 aumentan el riesgo de padecer lupus.[27]
Además existen por lo menos cinco regiones cromosómicas, independientemente del HLA, que contienen genes de predisposición, algunos asociados con la inmunidad innata (IRF5, STAT4, IRAK1, TNFAIP3, SPP1, TLR7), la mayoría asociados con la vía del interferón alfa; y otros involucrados en la señalización linfocitaria (PTPN22, OX40L, PD-1, BANK-1, LYN, BLK), que intervienen en la activación o supresión de los linfocitos T o B.[21][28]
Probablemente se requiera la presencia de genes de susceptibilidad, o la presencia de genes de susceptibilidad más la ausencia de genes protectores (como el polimorfismo TLR5 o la pérdida de función de la variante PTPN22) para conseguir la suficiente susceptibilidad genética y permitir el desarrollo de la enfermedad. Estas combinaciones genéticas modifican las respuestas inmunitarias al ambiente externo e interno; cuando dichas respuestas son excesivas o demasiado prolongadas aparecería la autoinmunidad. Adicionalmente, algunos de los polimorfismos en genes de riesgo de LES pueden predisponer a diversos subtipos en las manifestaciones clínicas del LES.[21][27]
Factores hormonales
[editar]El LES es mucho más frecuente en mujeres que en varones. Esto ha llevado a asumir que las hormonas tanto endógenas como exógenas (anticoncepción hormonal, terapia de sustitución hormonal) tienen un rol relevante en la producción y desarrollo de la enfermedad. Sin embargo, no existe consenso respecto de la forma y la importancia de su influencia (positiva o negativa) en este sentido.[29]
Las hembras de varias especies de mamíferos elaboran respuestas ante antígenos con mayor producción de anticuerpos que los machos.[cita requerida]
Existe evidencia de que el uso de estrógenos aumenta la posibilidad de desarrollar o producir un brote de LES, así como el uso de andrógenos tiene un efecto protector. Sin embargo, no hay evidencia clara de que el uso de anovulatorios orales con estrógenos u otro tipo de anticoncepción hormonal tenga efectos directos en este sentido. La terapia de reemplazo hormonal en mujeres postmenopáusicas pueden asociarse a una mayor frecuencia en la aparición de brotes de LES leves o moderados.[29][30]
Los estrógenos se han asociado a la estimulación de los linfocitos T y B, macrófagos y citocinas, amplificando su activación y supervivencia, con lo que se favorece una respuesta inmune más prolongada. Además de los estrógenos, existen evidencias sustanciales sobre las funciones inmunoreguladoras de otras hormonas como progesterona, testosterona, deshidroepiandrosterona y hormonas hipofisarias, incluyendo la prolactina. Estas observaciones han apoyado la hipótesis de que estas hormonas modulan la incidencia y severidad del LES.[31]
A pesar de los posibles efectos de las hormonas sexuales sobre el LES, la expresión clínica de la enfermedad es similar en mujeres y hombres, aunque los hombres en conjunto tienen un peor pronóstico general, con mayor daño orgánico, lo cual invalida cualquier hipótesis que apunte hacia los estrógenos como causa principal de la enfermedad.[21][31]
Factores ambientales
[editar]Se han propuesto varios factores ambientales que podrían intensificar o desencadenar un cuadro clínico de lupus, probablemente a través de sus efectos sobre el sistema inmune. Entre los factores de este tipo se incluye el tabaquismo, la dieta, la exposición a sílice, la exposición a la radiación ultravioleta, algunas infecciones y ciertos fármacos.
- La radiación ultravioleta puede exacerbar la sintomatología del LES, sin embargo, su rol en la patogenia de la enfermedad no está claro. Si bien varios estudios relacionan la fotosensibilidad y la aparición de brotes de LES con la exposición a esta radiación, no hay una recomendación concluyente respecto de su rol en la incidencia y desarrollo de la enfermedad.[32]
- Infecciones: los pacientes con LES tienen títulos más altos de anticuerpos para el virus de Epstein-Barr que los controles de edad, sexo y etnia similares sin LES, además de una mayor carga viral de este virus. Estudios en niños con LES sugieren que la infección por el virus de Epstein-Barr puede ser un desencadenante que origine el lupus clínico.[21] Este virus activa a los linfocitos B y además contiene secuencias de aminoácidos que simulan secuencias en los empalmosomas, especificidad común de los autoanticuerpos en las personas con LES, que puede contribuir al desarrollo de autoinmunidad por mimetismo molecular.[21] Otro tanto se ha descrito con los retrovirus.[33] Las infecciones por micobacterias y la tripanosomiasis pueden inducir anticuerpos anti-DNA o incluso síntomas de lupus, y las infecciones bacterianas pueden desencadenar brotes de la enfermedad.[21]
- Lupus eritematoso inducido por fármacos: ciertos fármacos pueden originar un aumento de la autoinmunidad, que en algunas personas acaba produciendo un cuadro clínico, variante del LES, denominado lupus eritematoso inducido por fármacos.[34] Hay cerca de 90 medicamentos actualmente en uso que pueden causar este cuadro, si bien los más comunes son los antiarrítmicos procainamida y quinidina, y el antihipertensivo hidralazina.[34][35] El cuadro consiste en la aparición de diversas manifestaciones clínicas, generalmente fiebre, malestar general, artralgias o artritis, mialgias intensas, serositis —pleuritis o pericarditis— y erupción cutánea, o cualquier combinación de estos datos, junto con anticuerpos antinucleares (ANA) positivos.[36][37] Es un estado reversible que desaparece cuando el paciente deja de tomar la medicación que desencadenó el episodio.
- Otros: el polvo de sílice, el uso de lápiz de labios y fumar cigarrillos pueden aumentar el riesgo de desarrollar LES.[38][39][40] No hay evidencias de asociación entre LES y el uso de tintes de pelo, pesticidas o consumo de alcohol.[21]
Patogenia
[editar]

Lo característico de la patogenia del LES es la respuesta inmune dirigida contra los antígenos nucleares endógenos. Los autoantígenos liberados por células apoptóticas son presentados por las células dendríticas a los linfocitos T iniciando su activación. Estos linfocitos interactúan mediante citocinas como la interleucina 10 e interleucina 23 y mediante moléculas de superficie como el CD40L y el CTLA-4, con los linfocitos B para que éstos produzcan los anticuerpos contra estos componentes propios (por lo tanto se producen autoanticuerpos). También existe un mecanismo de producción de autoanticuerpos por los linfocitos B, no mediada por linfocitos T, a través de señales de los receptores BCR (propios de los linfocitos B) y los receptores de tipo Toll o TLRs.[8]
Los fenómenos patogénicos clave son los siguientes:[8]
- Apoptosis: una de las alteraciones celulares más evidente en los pacientes con LES es un trastorno en el proceso de muerte celular denominado apoptosis, así como en el aclaramiento o eliminación de estas células apoptóticas. La expresión anómala de genes que regulan la apoptosis se ha asociado al desarrollo de enfermedades parecidas al lupus en modelos animales. En los humanos con LES se han demostrado trastornos de la regulación de la apoptosis en diferentes tipos de células, particularmente en linfocitos T y en la vía ligando Fas/FasL. En el lupus, durante la apoptosis, los autoantígenos sufren una notable redistribución, pasando a estar concentrados en las vesículas de la superficie de las células apoptóticas, con lo que determinantes antigénicos intracelulares, habitualmente ocultos, se hacen visibles para el sistema inmune. Además, existe un defecto en la fagocitosis y eliminación de las células apoptóticas y de los restos celulares, lo que origina la presencia mantenida de autoantígenos intracelulares, siendo así los restos apoptóticos una fuente de autoantígenos fundamental en el lupus.[41][42][43][44][45]
- Células B y T: en el LES se produce un aumento notable del número de células circulantes secretoras de inmunoglobulinas —hasta 50 veces—, junto con una activación selectiva de las células B, dirigidas contra un número determinado de antígenos.[46] Las células T son importantes en la activación de las células B y en la posterior producción de autoanticuerpos. Aunque en el LES el número total de células T está disminuido, se ha observado en estos pacientes la presencia de células T hiperactivas específicas para autoantígenos.[46] También se han descrito múltiples anomalías de señalización, tanto en los linfocitos T como en los B, incluyendo hiperactividad e hiperreactividad celular.[21][47] La hiperreacción de las células T y B se traduce en una mayor expresión de determinadas moléculas, como HLA-D y CD40L, lo que demuestra que las células son activadas fácilmente por los antígenos que inducen las primeras señales activadoras y las moléculas que estimulan la activación celular completa a través de la segunda señal. El resultado final de estas anormalidades es la producción sostenida de autoanticuerpos patógenos y la formación de inmunocomplejos que se adhieren a ciertos tejidos.[48]
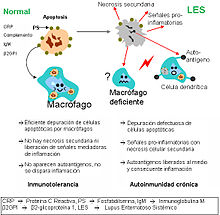
Los defectos en la depuración de células apoptóticas es una de las posibles explicaciones de ciertas enfermedades autoinmunes como el LES. - Autoanticuerpos: la producción de autoanticuerpos es un rasgo característico de los pacientes con LES. Su producción puede ser por activación policlonal de las células B o por estimulación inmune dirigida por autoantígenos.[47] Su importancia en el origen de la enfermedad se pone de manifiesto, por ejemplo, en el lupus eritematoso neonatal, que se piensa que es producido, al menos en parte, por el paso de los autoanticuerpos de la madre (anti-Ro y anti-La) al feto durante el embarazo, aunque también parece ser necesario un factor fetal complementario.[49] Además, los niveles del algunos autoanticuerpos llevan un curso paralelo a la actividad de la enfermedad y algunos se asocian con datos clínicos específicos de la enfermedad.[50]

- Autoanticuerpos e inmunocomplejos: los mediadores del LES son los autoanticuerpos y los inmunocomplejos que se forman con los antígenos. Los autoanticuerpos pueden estar presentes durante años antes de que aparezcan los primeros síntomas de la enfermedad.[51] Los autoantígenos que son reconocidos son presentados primariamente en la superficie de las células, particularmente por células activadas o bajo apoptosis, en las que los antígenos celulares han emigrado a la superficie celular, donde pueden ser reconocidas por el sistema inmune.[52] La fagocitosis y eliminación de los inmunocomplejos, de las células apoptóticas y de los restos derivados de las células necróticas son defectuosos en el LES, permitiendo la presencia de los antígenos y los inmunocomplejos durante períodos prolongados, haciendo que el daño en los tejidos vaya acumulándose hasta la aparición de la enfermedad clínica. Las células plasmáticas/células B que producen los autoanticuerpos están persistentemente activadas por el factor activador de células B (B lymphocyte stimulator o BLyS) y por las células T helper activadas, produciendo citoquinas como la IL-6 y IL-10.[21] También fallan en el LES los mecanismos de atenuación de estas funciones de las células T y B, incluyendo la generación de varios tipos de células T reguladoras y citolíticas y de sistemas atenuadores humorales idiotípicos.[48] Todos estos múltiples defectos originan una cascada de acontecimientos que comienzan con la anormal muerte celular y acaba en la célula B autoreactiva activada, proliferando y diferenciándose en células productoras de un exceso de autoanticuerpos frente a muchos antígenos nucleares. Además, y para complicar más la ya mala situación, se produce una activación del sistema inmune innato, con liberación de IL-1, TNF alfa, interferones, BLys y APRIL promoviendo la inflamación, además de la supervivencia de las células B autoreactivas. El resultado final es la producción de más anticuerpos antinucleares específicos, que pueden preceder a la manifestaciones clínicas durante años.[21]

- Lesión tisular: los autoanticuerpos pueden producir lesión tisular por el depósito de inmunocomplejos y la reacción inflamatoria secundaria o por interferir directamente en las funciones celulares.[47] No todos los autoanticuerpos causan la enfermedad. De hecho, todos los individuos normales hacen autoanticuerpos, aunque en pequeñas cantidades. La variabilidad en la enfermedad clínica que existe entre diferentes pacientes puede por tanto reflejar la variabilidad en la cantidad y calidad de la respuesta inmune, incluyendo su red reguladora.[21] Diversas características generales aumentan la probabilidad de que un autoanticuerpo sea patogénico, incluyendo su capacidad para unirse directamente a tejidos diana, su capacidad de activar el complemento, su carga catiónica, que favorece la adherencia a las membranas, o su avidez por el autoantígeno presente en el tejido diana.[46] Por su parte, algunos factores favorecen la patogenicidad de los inmunocomplejos, como las cantidades excesivas de inmunocomplejos —que superan los mecanismos de eliminación—, un tamaño correcto —los de tamaño intermedio son los que más posibilidades tienen de escapar a los mecanismos de eliminación—, un mayor tropismo tisular de los inmunocomplejos —debido a su carga catiónica—, o una disminución de los mecanismos de eliminación.[46]
- Aunque el lupus eritematoso sistémico ha sido etiquetado originalmente como una enfermedad mediada por immunocomplejos, cada vez hay más evidencias de que su patogénesis es más que eso, incluyendo otras interacciones complejas entre individuos predispuestos y su entorno.[44]
Cuadro clínico
[editar]| Síntoma | Inicio | Evolución |
|---|---|---|
| Síntomas constitucionales | 73 % | 84-86 % |
| Artralgias | 77 % | 84-86 % |
| Artritis | 56 % | 63-92 % |
| Miositis | 7 % | 5 % |
| Lesiones cutáneas | 57 % | 72-81 % |
| Lesiones mucosas | 18 % | 9-54 % |
| Fenómeno de Raynaud | 33 % | 18-58 % |
| Pleuritis | 23 % | 37-45 % |
| Afectación pulmonar | 9 % | 17 % |
| Pericarditis | 20 % | 29-31 % |
| Miocarditis | 1 % | 4-8 % |
| Afectación renal | 44 % | 46-77 % |
| Síndrome nefrótico | 5 % | 11-23 % |
| Alteraciones del sistema nervioso central | 24 % | 26-54 % |
| Afectación gastrointestinal | 22 % | 47-49 % |
| Pancreatitis | 1 % | 4 % |
| Adenopatías | 25 % | 32-59 % |
El LES comienza con una etapa preclínica, en la cual no hay síntomas, pero existen autoanticuerpos comunes al LES y a otras enfermedades inmunológicas. Luego comienzan a manifestarse distintos signos y síntomas, en forma muy variable tanto en órganos afectados como en intensidad; además el hecho de que su etapa clínica alterne períodos de remisión y recidivas, hacen que su diagnóstico sea especialmente difícil. En cualquier caso, el patrón clínico con que la enfermedad se presenta durante los primeros años tiende a prevalecer posteriormente.[4][8]
Manifestaciones generales
[editar]Los síntomas constitucionales, como malestar general, cansancio, fiebre, anorexia y pérdida de peso son comunes en los pacientes con LES, pudiendo ser las manifestaciones iniciales de la enfermedad o ser debidos a complicaciones de la misma.[54] El cansancio es frecuente y puede estar asociado al propio lupus, a anemia, hipotiroidismo, algunas medicaciones —como prednisona y beta bloqueantes—, trastornos del sueño, síndrome de fibromialgia o factores psicológicos.[55] La fiebre es un reto clínico en estos pacientes. Aproximadamente un 36 % de los pacientes debuta la enfermedad con fiebre y cerca de la mitad de los enfermos con LES tiene fiebre como manifestación de un lupus activo, pero también puede ser manifestación de otros problemas, en especial de infecciones.[4][6]
Manifestaciones músculo-esqueléticas
[editar]- Artritis: el 90 % de los pacientes con lupus presenta artritis —inflamaciones articulares—, siendo las articulaciones más afectadas las interfalángicas proximales y metacarpofalángicas de manos, muñecas y rodillas, aunque puede afectarse cualquier otra articulación. A diferencia de la artritis reumatoide, el lupus eritematoso sistémico no es una enfermedad que normalmente destruya el hueso, sin embargo, las deformidades causadas por la enfermedad pueden llegar a ser irreversibles en al menos el 20 % de los pacientes, pudiendo observarse deformidades en cuello de cisne y desviación cubital de las metacarpofalángicas de modo parecido a la artritis reumatoide.[56] Las erosiones son raras y en menos del 10 % se pueden observar nódulos subcutáneos, similares a los de la artritis reumatoide.[19]
- Dolores musculares: las mialgias y la hipersensibilidad muscular son frecuentes en las etapas activas de la enfermedad, especialmente en los niños. Entre un 5 a un 11 % de los pacientes con lupus presentan miositis inflamatorias en los músculos proximales de las extremidades.[6][8]
- Osteonecrosis: los pacientes con lupus tienen una prevalencia mayor que la población general de necrosis isquémica de los huesos, en especial en pacientes tratados con glucocorticoides. Se debe sospechar ante la aparición de dolor agudo en una sola articulación, como cadera, rodilla y hombro.[56]
Manifestaciones cutáneas
[editar]
Las lesiones cutáneas aparecen en un 80 % de los pacientes con LES y forman parte de los criterios de clasificación de la enfermedad. En concreto, en los criterios de 2012, son criterios las lesiones de lupus cutáneo agudo o subagudo, las lesiones de lupus crónico, la alopecia y las úlceras orales o nasales. En los criterios de 1997 lo era también la fotosensibilidad.
Las lesiones cutáneas específicas más frecuentes son:
- Lupus cutáneo agudo: la lesión más reconocida es la erupción malar o eritema en alas de mariposa, presente en el 30-50 % de pacientes. La erupción malar es una erupción eritematosa fija, plana o elevada que afecta a mejillas y puentes de la nariz, y que con frecuencia afecta a la barbilla y pabellones auriculares. La erupción respeta el surco nasolabial y suele fluctuar con la actividad de la enfermedad.[19]
- Lupus cutáneo subagudo: el término se refiere a distintas lesiones cutáneas, en general pápulas o placas, más difusas, fotosensibles, no induradas y que no dejan cicatriz. Un 90 % de los pacientes con este tipo de afectación cutánea tiene anticuerpos anti-Ro.[47][57]
- Lupus crónico: el lupus discoide crónico es la forma más frecuente. Consiste en pápulas o placas eritematosas y descamativas o hiperqueratósicas, bien delimitadas, con tendencia a la cronicidad y crecimiento periférico, que dejan cicatrices atróficas, con pérdida de anexos y alteraciones de la pigmentación. Es la lesión cutánea lúpica que menos se relaciona con la afectación orgánica. Entre las formas crónicas también está el lupus eritematoso discoide hipertrófico, el lupus eritematoso tumidus, la perniosis lúpica y la paniculitis lúpica o lupus eritematoso profundo.[58]
Entre las manifestaciones cutáneas inespecíficas de la enfermedad destacan la fotosensibilidad, la alopecia no cicatricial, Fenómeno de Raynaud —ocurre en más del 50 %— y las úlceras orales y nasales. Son frecuentes en los pacientes con lupus y se relacionan más con la actividad de la enfermedad. También es posible encontrar, nódulos subcutáneos, lesiones ampollares, urticariformes, vasculíticas e infartos periungueales.[58]
Manifestaciones renales
[editar]Se encuentra afectación renal clínica aproximadamente un 50 % de los pacientes; sin embargo, la mayoría del resto de pacientes tiene enfermedad subclínica que se puede observar si se realiza una biopsia renal. La afectación renal habitualmente ocurre en los primeros años de la enfermedad y debería ser detectada de modo temprano mediante análisis periódicos de orina y de la función renal.[59] Una hematuria o proteinuria indoloras suelen ser el único hallazgo inicial renal. La afectación renal es una de las principales causas de morbilidad y mortalidad en LES; aunque, debido al reconocimiento y tratamiento precoz, las últimas fases de enfermedad renal crónica se observan en menos del 5 % de los paciente. Pueden presentarse diversas formas de glomerulonefritis, siendo la biopsia renal necesaria para determinar el tipo y la extensión de la afectación renal.[59]

En 2004 se desarrolló un sistema de clasificación de la nefritis lúpica, denominado clasificación ISN (International Society of Nephrology) determinada por la biopsia renal.[60]
- Nefritis lúpica con cambios mínimos mesangiales (clase I).
- Nefritis lúpica con proliferación mesangial (clase II).
- Nefritis lúpica focal (clase III).
- Nefritis lúpica difusa (clase IV).
- Nefritis lúpica membranosa (clase V).
- Nefritis lúpica esclerosante evolucionada (clase VI).
Además de las lesiones glomerulares hay otras formas de enfermedad renal en el LES, incluyendo nefritis tubulointersticial, enfermedad vascular, microangiopatía trombótica y lupus podocitopatía.[61]
Manifestaciones neurológicas
[editar]Se trata del Neuropsychiatric systemic lupus erythematosus o NPSLE, en francés neurolupus. Las complicaciones neurológicas del LES incluyen disfunción cognitiva, síndrome cerebral orgánico —un estado confusional agudo—, delirio, psicosis, crisis convulsivas de cualquier tipo, cefalea —migrañosa o inespecífica—, corea y neuropatías periféricas. Otros problemas neurológicos menos frecuentes son trastornos del movimiento, neuropatías craneales, mielitis y meningitis. Las alteraciones del sistema nervioso central son en algunos casos la causa principal de morbilidad y mortalidad.[62]
- La disfunción cognitiva es la manifestación más frecuente del lupus difuso del sistema nervioso central, en particular problemas de memoria y razonamiento.[59]
- El LES puede cursar con convulsiones de cualquier tipo. Las crisis comiciales suelen ser de aparición temprana en la evolución de la enfermedad. Su tratamiento casi siempre exige un anticomicial y un inmunosupresor.[62]
- Pueden ocurrir cuadros tromboembólicos cerebrales, a menudo asociados a anticuerpos antifosfolípidos o con anticoagulante lúpico, aproximadamente en un 20 % de pacientes. Estos cuadros pueden causar ictus o crisis convulsivas o defectos cognitivos más difusos.[59]
- La neuropatía periférica se presenta hasta en un 18 % de los pacientes. Lo más común es una polineuropatía sensoriomotora de predominio distal; sin embargo, también puede expresarse como déficit fragmentarios o mononeuritis múltiple o una polineuropatía desmielinizante aguda. El cuadro clínico puede confundirse con una miastenia gravis o un síndrome miasténico de Lambert-Eaton.[63]
- El compromiso de pares craneales se observan en un 10 % de pacientes con LES. En cuanto a los nervios craneales los más afectados son los nervios sensitivos y motores del ojo y el trigémino. No obstante, cualquier par craneal puede verse afectado tanto en forma unilateral como bilateral.[63]
- La psicosis, que puede ser debida al lupus o al tratamiento con corticoides, es una de las manifestaciones neuropsiquiátricas más severas del LES. En ocasiones puede ser la manifestación dominante del lupus. Otras alteraciones psiquiátricas incluyen ansiedad, depresión y manía.[62]
Manifestaciones pulmonares
[editar]En el LES se puede observar pleuritis, con o sin derrame pleural, neumonitis intersticial, hipertensión pulmonar y hemorragia alveolar. Además, el riesgo de acontecimientos tromboembólicos está aumentado en los pacientes con anticuerpos antifosfolípidos.[59]
La tos se asocia la mayoría de las veces, con infección respiratoria alta de etiología vírica; sin embargo, la afectación pulmonar en el lupus debe ser objeto de evaluación cuidadosa, para descartar neumonitis lúpica aguda, caracterizada por pleuresía, disnea, tos y fiebre, con infiltrados pulmonares en las radiografías, que precisa tratamiento inmunosupresor y cuidados respiratorios de soporte.[19]
La presencia de disnea, dolor torácico pleurítico episódico y disminución progresiva del volumen pulmonar en ausencia de fibrosis intersticial o enfermedad significativa de la pleura sugiere la presencia del síndrome del pulmón encogido.[19]
Manifestaciones cardíacas
[editar]
Existe una gran variedad de manifestaciones cardiovasculares del LES. Los pacientes con lupus pueden presentar inflamación en todas las partes del corazón, originándose pericarditis, miocarditis o endocarditis.
- La pericarditis es relativamente común en los pacientes con lupus. Los derrames pericárdicos pueden progresar rápidamente junto con la actividad general del lupus. Estos derrames suelen responder a dosis altas de corticoides.[19]
- La endocarditis del lupus eritematoso sistémico es característicamente no infecciosa, denominada endocarditis de Libman-Sacks, e implica tanto a la válvula mitral como a la válvula tricúspide; aunque habitualmente es silente, puede producir insuficiencia valvular y puede servir de fuente de émbolos.[59]
- La miocarditis es poco frecuente, pero puede ser severa.[59]
Los pacientes con LES tiene un riesgo aumentado de arteriosclerosis precoz y de enfermedad arterial coronaria, casi siempre por arteriosclerosis acelerada, aunque también puede ser por vasculitis.[59]
Manifestaciones gastrointestinales
[editar]El compromiso gastrointestinal del LES es poco frecuente. Suelen presentarse trastornos gastrointestinales debido a los efectos adversos de la medicación (AINE y corticoides) que por el lupus activo. Es frecuente la presencia de dolor abdominal inespecífico. Cualquier área del tubo digestivo puede estar afectado por el LES o sus complicaciones. Algunas exacerbaciones del LES se manifiestan por náuseas, a veces acompañadas de vómitos y dolor abdominal difuso, debido a una peritonitis autoinmune o lúpica. La peritonitis constituye hasta el 30 % de las serositis en los pacientes con LES.[6][4]
En las fases de actividad de la enfermedad es habitual encontrar elevación de las enzimas hepáticas —GOT y GPT— que se normalizan con el tratamiento y control de la enfermedad. En raras ocasiones se desarrolla una hepatitis crónica activa, siendo otras manifestaciones poco frecuentes la pancreatitis, cirrosis biliar primaria, colangitis autoinmune, hiperplasia nodular regenerativa y vasculopatía oclusivo trombótica con síndrome de Budd-Chiari.[48][47]
La vasculitis intestinal, con dolor abdominal agudo, vómitos y diarrea, puede ser un cuadro grave; siendo sus complicaciones la perforación, isquemia, hemorragia intestinal y sepsis.
Manifestaciones hematológicas
[editar]Las citopenias —disminución de algún tipo de células sanguíneas— y trombofilia, —con una propensión a desarrollar cuadros tromboembólicos— pueden ser datos clínicos del LES.
- Citopenias
- Los pacientes con lupus desarrollan con frecuencia alteraciones en uno o más de los tres tipos de células sanguíneas: trombocitopenia, anemia, leucopenia.
- Trombosis: es la más frecuente; sin embargo, no suele producirse sangrado salvo cuando las cifras de plaquetas son inferiores a 25 000/mm³. La asociación de anemia hemolítica y trombopenia recibe el nombre de síndrome de Evans.[47]
- Anemia: muchos pacientes tienen anemia, que habitualmente es debida a anemia de trastorno crónico. La anemia hemolítica que se presenta en un 10 % puede ser severa. Otras formas de anemia en el LES son la anemia por insuficiencia renal, la anemia microangiopática, la anemia ferropénica y la aplasia inducida por fármacos.[47]
- Leucopenia: la leucopenia es muy común, encontrándose recuentos de leucocitos por debajo de 4500/mm³ en más del 50 % de pacientes. Aunque tiene utilidad en el diagnóstico, raramente da problemas, salvo que sea severa, con recuentos inferiores a 2000/mm³. Se relaciona con la actividad de la enfermedad y suele ir asociada a linfopenia —disminución del número de linfocitos—.
- Trombofilia
- Algunos pacientes con LES, en especial aquellos con anticuerpos antifosfolípidos o con síndrome nefrótico severo, tienen un riesgo aumentado de enfermedad tromboembólica, que se puede manifestar como trombosis venosa o como enfermedad arterial.[59]
- Adenopatías y esplenomegalia
- Muchos pacientes tienen adenopatías periféricas o esplenomegalia.
Existen dos síndromes caracterizados por citopenias y trastornos de la coagulación que se pueden asociar al LES. Cuando son la manifestación inicial, puede ser difícil discernir si se trata de una manifestación más en el contexto del lupus o un cuadro independiente:
- Síndrome antifosfolípido: el síndrome antifosfolípido, descrito inicialmente en conexión con el LES, se observa en un 20-35 % de los pacientes, siendo independiente de la actividad y severidad de la enfermedad. Se caracteriza por trombosis arterial y venosa, abortos recurrentes, citopenias y enfermedad neurológica, junto con la presencia de anticuerpos antifosfolípidos —anticoagulante lúpico, anticuerpos anticardiolipina y anti-β2-glicoproteína I—.[64] La presencia de este síndrome empeora el pronóstico de los pacientes con LES.[65]
- Síndrome hemofagocítico: el síndrome hemofagocítico se ha descrito asociado al LES y, en ocasiones, como forma de presentación de la enfermedad. Se trata de un cuadro con fiebre mantenida, pancitopenia progresiva, organomegalia, disfunción hepática, coagulopatía y aumento notable de la ferritina. El dato definitivo es la presencia de células macrofágicas fagocitando células hematopoyéticas en médula ósea, bazo o ganglios linfáticos.[66]
Otras alteraciones
[editar]- Alteraciones endocrinas: se ha descrito hiperprolactinemia hasta en el 20 % de los pacientes de ambos sexos. Las pruebas de función tiroidea son anormales con frecuencia, siendo más prevalente en los pacientes con LES el hipotiroidismo. También se han descrito vasculitis en los ovarios y mayor frecuencia de anomalías en los espermatozoides en los pacientes con LES.[cita requerida]
- Afectación ocular: el síndrome seco y cambios vasculares retinianos por vasculitis retiniana —con observación de exudados como granos de algodón o cuerpos citoides— o por trombosis microvascular, son los hallazgos más frecuentes.[53] También se pueden observar queratitis ulcerativa periférica, coroidopatía, uveitis y neuritis óptica. Otras alteraciones como glaucoma y catarata son secundarias al tratamiento con corticoides, y la maculopatía como complicación del tratamiento con antipalúdicos.[67]
- Oído, nariz y laringe: se han descrito sordera neurosensorial y afectación laríngea por cricoaritenoiditis y parálisis de cuerdas vocales. En la mucosa de la boca y nariz es frecuente la aparición de úlceras. Ocasionalmente puede ocurrir perforación del tabique nasal, como resultado de vasculitis.[53]
Diagnóstico
[editar]Los pacientes con LES suelen presentar con datos clínicos muy variables, desde una enfermedad articular y cutánea leves, hasta un cuadro grave, con afectación renal, hematológica o del sistema nervioso central y con riesgo para la vida del paciente.[68] Por otra parte, no existe una prueba única e inequívoca para el diagnóstico del lupus, lo que unido a la variabilidad clínica de esta enfermedad, hace que el diagnóstico sea con frecuencia un reto para el médico.[68]
Pruebas de laboratorio
[editar]
Los estudio de laboratorio en los pacientes con LES ayudan a establecer el diagnóstico, a controlar la evolución de la enfermedad y a identificar posibles complicaciones de la misma o efectos adversos de las medicaciones.
Las alteraciones hematológicas, sobre todo la leucopenia, linfopenia y trombopenia, son frecuentes en el LES. La VSG suele estar elevada cuando la enfermedad está en actividad, mientras que la proteína C reactiva suele ser normal.[47]
La presencia de autoanticuerpos es la característica más prominente del LES. Los anticuerpos antinucleares (ANA) están presentes en más del 95 % de los pacientes, y su ausencia hace dudar del diagnóstico. Menos del 5 % de los pacientes con LES tienen ANA negativos cuando son detectados por inmunofluorescencia, siendo los porcentajes aún menores según el tipo de sustrato utilizado y de la técnica empleada para su detección. Los patrones de inmunofluorescencia más habituales son el homogéneo, el moteado y el periférico, y los títulos suelen ser elevados.[48] Los anticuerpos anti DNA bicatenario (anti-DNAn) son muy específicos del LES y se relacionan con la actividad de la enfermedad y la presencia de nefritis. La presencia de un patrón moteado en la inmunofluorescencia sugiere la presencia de anticuerpos dirigidos frente a antígenos extraíbles del núcleo (ENA). Entre estos se encuentran el anti-Sm —también muy específico del LES—, anti-RNP, anti-Ro y anti-La. Los anticuerpos anti-Ro indican una mayor riesgo de padecer lupus cutáneo subagudo, lupus neonatal y síndrome de Sjögren.[48]
El factor reumatoide puede estar presente en un 40 % de los pacientes. La actividad hemolítica del complemento está disminuida y se correlaciona también con el grado de actividad de la enfermedad. Los componentes C3 y C4 son los que presentan actividad más baja.
En las pruebas de coagulación, es frecuente el alargamiento del tiempo parcial de tromboplastina activada y sugiere la presencia de anticuerpos antifosfolípidos, incluyendo anticoagulante lúpico, anticuerpos anticardiolipina y anti β2-glicoproteína. Un 25 % de los pacientes con LES tienen un test falsamente positivo para la sífilis, traduciendo, al igual que el alargamiento del tiempo parcial de tromboplastina, la presencia de anticuerpos antifosfolípidos.[47]
El análisis sistemático de orina es fundamental para vigilar la aparición de nefritis, evidenciándose en este caso la presencia de hematuria, leucocituria, cilindros celulares y grados variables de proteinuria.[47]
En ausencia de unos criterios diagnósticos de LES, los médicos utilizan habitualmente los criterios de clasificación de LES como guía para ayudar a identificar algunos de los datos clínicos más destacados cuando hacen el diagnóstico. La prueba de los anticuerpos antinucleares (ANA) es positiva virtualmente en todos los pacientes con LES en algún momento en el curso de su enfermedad y algunos anticuerpos, como los anti-DNAn y anti-Sm son muy específicos de LES, estando presentes en aproximadamente el 70 % y 30 % de pacientes con LES respectivamente.[68]
Criterios de clasificación del LES
[editar]| Criterios del ACR de 1982[69] revisados en 1997[70] para la clasificación de LES |
|---|
| 1. Erupción malar. |
| 2. Lupus discoide crónico. |
| 3. Fotosensibilidad. |
| 4. Úlceras orales o nasofaríngeas. |
| 5. Artritis no erosiva, afectando a dos o más articulaciones periféricas. |
| 6. Pleuritis o pericarditis. |
| 7. Afectación renal: proteinuria (>500 mg/24 horas o > 3+) o cilindros celulares —hematíes, hemoglobina, granulares, tubulares o mixtos— en el sedimento. |
| 8. Afectación neurológica: convulsiones o psicosis. |
| 9. Alteraciones hematológicas: anemia hemolítica con reticulocitosis o leucopenia (<4000 mm³, en dos ocasiones) o linfopenia (<1500 mm³ en dos ocasiones) o trombopenia (<100 000 mm³ en dos ocasiones). |
| 10. Alteraciones inmunológicas: anti-DNAn + o anti-Sm + o antifosfolípidos + (anticuerpos anticardiolipina IgG o IgM o anticoagulante lúpico o falso reactante de lues). |
| 11. Anticuerpos antinucleares +. |
| Se necesitan 4 de los 11 criterios para clasificar a un paciente como afectado por lupus eritematoso sistémico. |
Se han desarrollado varios criterios de clasificación para el LES con el fin de estandarizar a los pacientes para la realización de estudios. Con frecuencia se hace el diagnóstico de LES siguiendo estos criterios de clasificación, sin embargo, algunos pacientes pueden tener un lupus eritematoso sistémico a pesar de no haber cumplido nunca estos criterios de clasificación. Estos criterios pueden ser utilizados para documentar de modo sistemático los datos clínicos clave de la enfermedad, aunque tienen limitaciones en su uso con fines diagnósticos.[68]
- Los criterios ACR de 1997
- En 1971 la American Rheumatism Association (ARA) presentó unos criterios clínicos destinados a facilitar la comparación de los pacientes en los estudios epidemiológicos y en los ensayos clínicos.[71] Estos criterios fueron revisados en 1982 por el American College of Rheumatology (ACR) retirando algunos de los previos —Raynaud, alopecia y células LE— e introduciendo los ANA.[69] En 1997 se revisaron los criterios de 1982, añadiendo alguno de los avances en el conocimiento de la enfermedad producidos en los años previos, como los anticuerpos antifosfolípidos.[70] Según estos criterios un paciente puede ser clasificado como LES si presenta cuatro o más de los 11 criterios, bien simultáneamente o bien a lo largo del periodo de observación. Cuando fueron probados frente a otras enfermedades reumáticas, estos criterios tenían una sensibilidad y especificidad del 96 % aproximadamente.[68] Una de las mayores limitaciones de estos criterios era que los pacientes con nefritis lúpica confirmada por biopsia podían no cumplir los criterios y, por otra parte, pacientes que solo presentaban manifestaciones cutáneas podían cumplir los criterios de lupus sistémico. Además, los criterios ACR incluían la posible duplicación de criterios cutáneos muy relacionados —como el exantema malar y la fotosensibilidad—, y en ellos faltaba la inclusión de otras manifestaciones cutáneas, neurológicas e inmunológicas también características.[68][72]
- Criterios SLICC de 2012[73]
- En 2012 el grupo de trabajo SLICC (The Systemic Lupus International Collaborating Clinics) propuso una revisión de los criterios de clasificación para mejorar las debilidades de los criterios del ACR de 1997. Los criterios de clasificación de 2012 requieren la presencia de al menos 4 de los 17 criterios, incluyendo al menos 1 de los 11 criterios clínicos y 1 de los 6 criterios inmunológicos, o que el paciente tenga una nefritis lúpica comprobada por biopsia y la presencia de anticuerpos antinucleares (ANA) o anti-DNAn. Estos criterios fueron validados en pacientes con LES, teniendo una mayor sensibilidad y algo menor especificidad que los criterios ACR de 1997.[68] Sin embargo, pese a este aumento de la sensibilidad, todavía estos criterios pueden retrasar el diagnóstico en algunos pacientes y algunos no pueden clasificarse como lupus, por lo que algunos autores opinan que se podría diagnosticar a un paciente de LES teniendo 2 o 3 criterios del ACR o SLICC —en especial si algunos de ellos es muy específico, como los anti-DNAn o anti-Sm— y al menos otro dato clínico relevante, como neuritis óptica, meningitis aséptica, hematuria glomerular, neumonitis, hemorragia pulmonar, hipertensión pulmonar, enfermedad pulmonar intersticial, miocarditis, endocarditis de Libman-Sacks, vasculitis abdominal o fenómeno de Raynaud.[68]
| Criterios de 2012 para la clasificación del lupus eritematoso sistémico[73] | ||
|---|---|---|
| Criterio | Definiciones | |
| Criterios clínicos | 1. Lupus cutáneo agudo: exantema malar, lupus bulloso, exantema maculopapular lúpico, exantema lúpico fotosensible (en ausencia de dermatomiositis) o necrosis tóxica epidérmica variante de LES o lupus cutáneo subagudo. 2. Lupus cutáneo crónico: lupus discoide clásico, lupus hipertrófico (verrugoso), paniculitis lúpica, lupus eritematoso tumidus o perniosis lúpica. 3. Úlceras orales o nasales: en ausencia de otras causas como artritis reactiva, Behçet, vasculitis... 4. Alopecia no cicatricial: fragilidad o adelgazamiento capilar, en ausencia de otras causas. 5. Afectación articular: inflamación en 2 o más articulaciones, o dolor a la presión en 2 o más articulaciones + rigidez matinal >30 minutos. 6. Serositis: pleuritis o pericarditis de más de un día de duración, en ausencia de otras causas. 7. Afectación renal: relación proteinuria/creatinuria (o proteínas en orina de 24 horas) que representen 500 mg de proteínas/24 horas o cilindros hemáticos. 8. Alteraciones neurológicas: convulsiones, psicosis, mononeuritis múltiple, mielitis, neuropatía periférica o craneal, estado confusional agudo. 9. Anemia hemolítica. 10. Leucopenia o linfopenia: Leucocitos < 4000/mm³, al menos una vez, o linfocitos <1 000/mm³, al menos una vez, en ausencia de otras causas. 11. Trombocitopenia: Plaquetas <100 000/mm³ en ausencia de otras causas. | |
| Criterios serológicos | 1. ANA +, en valores por encima del nivel de referencia del laboratorio. 2. AntiDNAn +, en valores por encima del nivel de referencia del laboratorio (o >2 veces el nivel de referencia si es realizado por ELISA). 3. Anti Sm +. 4. Anticuerpos antifosfolípidos: anticoagulante lúpico, falso reactante de reagina, títulos medios o altos de anticardiolipina (IgA, IgG o IgM), resultado + para anti ß²-glicoproteína (IgA, IgG o IgM). 5. Complemento bajo: C3, C4 o CH50 bajos. 6. Coombs directo +. | |
| Los criterios son acumulativos y pueden no estar presentes simultáneamente. El paciente debe cumplir al menos 4 criterios, incluyendo al menos un criterio clínico y uno inmunológico o el paciente debe tener una nefritis lúpica comprobada por biopsia con ANA o anti-DNAn positivos. | ||
Comorbilidad
[editar]Los pacientes con lupus eritematosos sistémico padecen una serie de afecciones asociadas con mayor incidencia que la población general, incluyendo infecciones, arteriosclerosis, enfermedad cardiovascular, osteoporosis, cáncer[74] y enfermedad celíaca[75][76] (con la cual comparten genética).[77]
Su prevención y tratamiento mejora notablemente el pronóstico general de la enfermedad y la calidad de vida de los pacientes.[78]
Infecciones
[editar]- La susceptibilidad a las infecciones en los pacientes con LES está relacionada con el grado de actividad de la enfermedad y con los tratamientos esteroideo e inmunosupresor.[79] Otras alteraciones, como los déficit de complemento congénitos o adquiridos, también aumentan la susceptibilidad a las infecciones. Las localizaciones más frecuentes de las infecciones son el aparato respiratorio, el urinario y la piel.[74]
- Arteriosclerosis prematura
- La arteriosclerosis tiene una prevalencia aumentada y un desarrollo acelerado en los pacientes con LES.[80] En su patogenia intervienen los factores de riesgo tradicionales, como hipertensión, colesterol elevado, fumar, diabetes…, así como otros dependientes de la enfermedad —la propia actividad de la enfermedad— y el tratamiento prolongado con corticoides.[74]
- Afectación cardiovascular
- La enfermedad coronaria prematura cada vez es más reconocida como una causa de muerte en las fases tardías de la enfermedad. El riesgo de enfermedad coronaria o de accidente cerebrovascular es siete veces mayor en los pacientes con LES que en la población general.[81]
- Osteoporosis
- Entre los factores de riesgo de una mayor incidencia de osteoporosis en los pacientes con LES se citan a la actividad inflamatoria de la enfermedad, la escasa exposición solar, la posible menopausia precoz por el uso de inmunosupresores y, sobre todo, el tratamiento crónico con corticoides.[74]
- Cáncer
- Aunque los datos de la asociación del LES con tumores malignos han sido conflictivos, cada vez es más claro el riesgo aumentado de algunos tipos de tumores en estos pacientes, incluyendo linfomas, cáncer de pulmón, cáncer de mama y cáncer cervical. El uso de fármacos antipalúdicos parece estar asociado con un menor riesgo de cáncer.[79]
- Otras
- Otras comorbilidades en los pacientes con LES incluyen, síndrome de fibromialgia, hipertensión, diabetes mellitus y asociación a otras enfermedades autoinmunes.[74]
Tratamiento
[editar]Los pacientes con LES tienen una gran variabilidad clínica, siendo diferente el tratamiento según la afectación de órganos y sistemas que presenten y la gravedad de estas manifestaciones.
Los síntomas de la enfermedad suelen tratarse con antiinflamatorios no esteroideos (AINE), corticoides, antipalúdicos de síntesis, inmunosupresores y, en los últimos años, con fármacos biológicos.[48]
Medidas generales
[editar]- Protección solar
- los pacientes con LES deben evitar la exposición solar, así como a otras fuentes de luz ultravioleta. Deben usar cremas de protección solar, preferiblemente las que bloquean tanto los UV-A, como los UV-B, con un elevado factor de protección (>50+).[79][82]
- Dieta y nutrición
- Existen pocos datos en relación con la utilidad de modificaciones dietéticas en los pacientes con LES. Se recomienda una dieta equilibrada, restricción de la sal si existe hipertensión o nefritis y adelgazar si existe sobrepeso. Es posible que los niveles de vitamina D sean bajos en los pacientes con lupus, debido en parte a la baja exposición solar, siendo necesario en estos casos suplementos de vitamina D.[82]
- Ejercicio
- La inactividad producida por la enfermedad activa origina una pérdida de masa muscular, desmineralización ósea y pérdida de fortaleza física, originando sensación de cansancio. Esto puede ser evitado con ejercicios graduales, adaptados a las posibilidades del paciente.[82]
- No fumar
- Fumar cigarrillos puede aumentar el riesgo de LES y de enfermedad más activa. Es aconsejable también dejar de fumar para evitar comorbilidades como arteriosclerosis acelerada, trombosis, osteoporosis y cáncer.[82]
Tratamientos específicos
[editar]
El patrón y la gravedad de la afectación orgánica determinará el tipo específico de fármacos utilizados para su tratamiento.
- AINE
- Los antiinflamatorios no esteroideos (AINE) generalmente son eficaces para las quejas musculoesqueléticas —artralgias, artritis—, la fiebre y las serositis —pleuritis, pericarditis— leves.[48]
- Antipalúdicos
- Los antipalúdicos de síntesis, como la hidroxicloroquina, se utilizan principalmente para las manifestaciones cutáneas y articulares, así como para el control general de la enfermedad.[79] La terapia con hidroxicloroquina debería darse a todos los pacientes con LES que no tengan contraindicaciones para este medicamento.[83]
- Corticoides
- Los corticoides tópicos son usados con frecuencia en las manifestaciones cutáneas del LES. Los corticoides sistémicos en dosis altas (1-2 mg/kg/día de prednisona) son utilizados solos o en combinación con inmunosupresores para los pacientes con afectación orgánica importante, particularmente la afectación renal y del sistema nervioso central, aunque también en casos de trombopenia severa, anemia hemolítica o afectación cardiopulmonar. Dosis más bajas (hasta 15 mg/día de prednisona) se utilizan a veces para síntomas constitucionales, cutáneos y articulares hasta que hagan su efecto los antipalúdicos u otros fármacos ahorradores de corticoides.[48]
- Inmunosupresores
- Los inmunosupresores como metotrexato, azatioprina, ciclofosfamida o micofenolato, se reservan para aquellos pacientes con afectación importante de algún órgano, o aquellos que no responden adecuadamente a los corticoides. La ciclofosfamida, el micofenolato o la azatioprina se administran junto con corticoides en los paciente con nefritis lúpica. La ciclofosfamida también se administra a los pacientes con afectación del sistema nervioso central, así como en caso de vasculitis sistémica y hemorragia alveolar.[79]
- Fármacos biológicos
- El belimumab —anticuerpo monoclonal que inhibe la forma soluble del estimulador de linfocitos B o BlyS— es el primer fármaco biológico aprobado para el LES. Se usa para el tratamiento de los pacientes con LES activo pese a estar recibiendo terapia estándar con AINE, corticoides, antipalúdicos o inmunosupresores; sin embargo, no ha sido evaluado en pacientes con nefritis lúpica activa severa ni en los pacientes con enfermedad activa del sistema nervioso central.[84] El rituximab —anticuerpo monoclonal contra linfocitos B— puede ser beneficioso para pacientes con manifestaciones de la enfermedad resistentes a otras terapias.[84]
Otros fármacos inmunosupresores y biológicos están siendo investigados, así como otras modalidades de tratamiento incluyendo el trasplante de células madres hematopoyéticas y la inmunoablación aislada —sin trasplante de células madre—, aféresis —plasmaféresis, leucoplasmaféresis, crioféresis— y las gammaglobulinas intravenosas.[79][85]
Pronóstico
[editar]El lupus eritematoso sistémico tiene un curso clínico heterogéneo, variando desde formas clínicas relativamente benignas hasta cuadros severos con fallo orgánico y muerte. La mayoría experimenta exacerbaciones que se alternan con períodos de remisión relativa; no obstante, es raro que la enfermedad remita completa y permanentemente —ausencia de síntomas sin tratamiento—.[79]
La tasa de supervivencia de los pacientes con LES ha mejorado drásticamente en los últimos sesenta años. En la década de los 50, muchos de los pacientes diagnosticados de LES (40 %) vivían menos de cinco años. Los avances en el diagnóstico y tratamiento han aumentado la supervivencia hasta el punto de que más del 90 % de los pacientes sobrevive ahora más de diez años y muchos pueden vivir con normalidad, sin apenas síntomas. Es importante hacer notar que en esta estadística (10 años) no indica un tiempo de vida media, sino simplemente la duración del estudio referido.
De acuerdo con la Fundación de Lupus de América, «hoy en día, la mayoría de la gente con lupus puede esperar vivir una vida normal».[86] La probabilidad de supervivencia varía en función de la afectación orgánica, siendo la mejor para las lesiones cutáneas y articulares y la peor para la afectación renal y del sistema nervioso central. El uso de fármacos antipalúdicos parece reducir las tasas de mortalidad.[87]
Las principales causas de muerte en los primeros años de la enfermedad están relacionadas con la actividad de la enfermedad —como afectación renal, cardiovascular o del sistema nervioso central—, o a infecciones debido a la inmunosupresión; mientras que las causas de muerte en etapas más tardías son debidas a complicaciones de la propia enfermedad —como insuficiencia renal—, a complicaciones del tratamiento —como infecciones—, a enfermedad cardiovascular y a tumores.[79] Son factores de mal pronóstico general para la supervivencia en LES, la presencia de enfermedad renal —especialmente la glomerulonefritis proliferativa difusa—, hipertensión, sexo masculino, edad joven, bajo estado socioeconómico, raza negra, presencia de anticuerpos antifosfolípidos y elevados índices de actividad de la enfermedad.[79]
Véase también
[editar]- Conectivopatía
- Enfermedades autoinmunes
- Lupus eritematoso cutáneo
- Lupus eritematoso neonatal
- Reumatología
Referencias
[editar]- ↑ a b c d e Garrick, Nancy (11 de abril de 2017). «Systemic Lupus Erythematosus (Lupus)». National Institute of Arthritis and Musculoskeletal and Skin Diseases (en inglés). Consultado el 11 de mayo de 2021.
- ↑ a b c d e f g Justiz Vaillant, Angel A.; Goyal, Amandeep; Bansal, Pankaj; Varacallo, Matthew (2021). «Systemic Lupus Erythematosus». StatPearls (en inglés) (StatPearls Publishing). PMID 30571026. Consultado el 11 de mayo de 2021.
- ↑ Danchenko, N; Satia, J A; Anthony, M S (2006-05-XX). «Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden». Lupus (en inglés) 15 (5): 308-318. ISSN 0961-2033. doi:10.1191/0961203306lu2305xx. Consultado el 11 de mayo de 2021.
- ↑ a b c d «Guía de Práctica Clínica sobre Lupus Eritematoso Sistémico». Guías de Práctica Clínica en el SNS. (Canarias: Ministerio de Sanidad, Servicios Sociales e Igualdad. Servicio de Evaluación del Servicio Canario de la Salud.). 2015. Consultado el 11 de febrero de 2017.
- ↑ Guía Clínica AUGE Lupus Eritematoso Sistémico. Santiago de Chile: Ministerio de Salud. 2013. Consultado el 11 de febrero de 2017.
- ↑ a b c d e Pedraz Penalva, T; Bernabeu Gonzálvez, P; Vela Casasempere, P (2013). «Lupus Eritematoso Sistémico». En Belmonte, Miguel A, ed. Enfermedades Reumáticas – Actualización SVR II Edición (2 edición). Sociedad Valenciana de Reumatología. Archivado desde el original el 31 de marzo de 2016. Consultado el 11 de febrero de 2017.
- ↑ a b Nobee, Alisa; Vaillant, Angel Justiz; Akpaka, Patrick Eberechi; Poon-king, Peter (2015). «Systemic Lupus Erythematosus (SLE): A 360 Degree Review» [Lupus eritematoso sitémico (LES): una revisión en 360 grados]. American Journal of Clinical Medicine Research (en inglés) (Science and Education Publishing) 3 (4): 60-63. doi:10.12691/ajcmr-3-4-1. Consultado el 11 de febrero de 2017.
- ↑ a b c d e Bertsias, George; Cervera, Ricard; Boumpas, Dimitrios T (mayo de 2015). «Systemic Lupus Erythematosus: Pathogenesis and Clinical Features» [Lupus eritematoso sitémico: patogenia y características clínicas]. En Bijlsma, Johannes WJ; Hachulla, Eric, eds. (en inglés) (2 edición). Reino Unido: INGRAM INTERNATIONAL INC. pp. 476-505. ISBN 9780727919243. Consultado el 13 de febrero de 2017.
- ↑ Wallace, Daniel J (diciembre de 2015). «Overview of the management and prognosis of systemic lupus erythematosus in adults» (en inglés). Consultado el 11 de febrero de 2017. (requiere suscripción).
- ↑ «Orígenes del Día Mundial del Lupus». Fundación Americana de Lupus y Federación Española de Lupus. Archivado desde el original el 15 de mayo de 2017. Consultado el 2 de mayo de 2015.
- ↑ a b c Wallace, Daniel J (2014). «The history of lupus» [La historia del lupus]. Lupus: The Essential Clinician's Guide [Lupus: la guía clínica esencial] (en inglés) (2ª edición). OUP USA. ISBN 0199361967. Consultado el 20 de febrero de 2017.
- ↑ a b c d e f g h Smith, CD; Cyr M, M (1988). «The history of lupus erythematosus. From Hippocrates to Osler» [La historia del lupus eritematoso. De Hipócrates a Osler]. Rheum Dis Clin North Am (en inglés) (Canadá) 14 (1): 1-14. PMID 3041483. Consultado el 20 de febrero de 2017.
- ↑ a b c d e f g h i j k l m n ñ o Mallavarapu, Ravi K; Grimsley, Edwin W (2007). «The History of Lupus Erythematosus» [La historia del lupus eritematoso]. South Med J (en inglés) (Lippincott Williams & Wilkins) 100 (9): 896-898. PMID 17902290. Consultado el 19 de febrero de 2017.
- ↑ Gómez-Puerta, José A; Cervera, Ricard (2008). «Lupus eritematoso sistémico». Medicina & Laboratorio, Volumen 14, Números 5-6, 2008 (Medellín, Colombia: Universidad de Antioquía) 14 (5-6): 211-223. Consultado el 20 de febrero de 2017.
- ↑ a b c d e Fonollosa Pla V, Vilardell Tarrés M. Antecedentes históricos y conceptos actuales. En: Lupus eritematoso sistémico (3.ª edición). Caduceo Multimedia S.L. (2009). ISBN 978-84-936320-2-1
- ↑ Harvey AM, Shulman LE, Tumulty PA, Conley CL, Schoenrich EH. Systemic lupus erythematosus: review of the literature and clinical analysis of 138 cases. Medicine (Baltimore). 1954;33(4):291-437. PMID 13223169
- ↑ Badillo-Tenorio, Rocío Elizabeth; Rivas-Larrauri, Francisco Eduardo (noviembre-diciembre de 2013). «Lupus eritematoso sistémico y gammaglobulina intravenosa». Acta Pediatr Mex 34 (6): 353-357. Consultado el 19 de febrero de 2017.
- ↑ a b c BenedeK TG. Historia de las enfermedades reumáticas. En: Compendio de las enfermedades reumáticas (10.ª ed). Arthritis Foundation (1993). ISBN 84-88313-61-6
- ↑ a b c d e f g Edworthy SM. Manifestaciones clínicas del lupus eritematoso sitémico. En: Kelley Tratado de reumatología (7.ª edición). Harris ED, ed. Elsevier España (2006). ISBN 978-84-8174-840-6
- ↑ Espinosa G, Cervera R. Epidemiología. En: Lupus eritematoso sistémico (3.ª edición). Caduceo Multimedia S.L. (2009). ISBN 978-84-936320-2-1
- ↑ a b c d e f g h i j k l m n ñ o p Schur PH, Hahn BH. Epidemiology and pathogenesis of systemic lupus erthematosus. UpToDate. 2014
- ↑ Lahita RG. The role of sex hormones in systemic lupus erythematosus. Curr Opin Rheumatol 199;11:352
- ↑ is lupus? Lupus Foundation
- ↑ Symmons DP. Frequency of lupus in people of African origin. Lupus. 1995;4(3):176-8. PMID 7655486
- ↑ Lalani S, Pope J, de Leon F, Peschken C; Members of CaNIOS/1000 Faces of Lupus. Clinical features and prognosis of late-onset systemic lupus erythematosus: results from the 1000 faces of lupus study. J Rheumatol. 2010;37(1):38-44. doi: 10.3899/jrheum.080957. PMID 20008925
- ↑ Rueda B, Orozco G, Sánchez E, Oliver J y Martín J. Factores genéticos comunes en autoinmunidad. Reumatol Clin 2008;4 Supl 1:S1-4. doi: 10.1016/S1699-258X(08)76131-X http://www.reumatologiaclinica.org/es/factores-geneticos-comunes-autoinmunidad/articulo/13117214/ Archivado el 17 de septiembre de 2018 en Wayback Machine.
- ↑ a b Ramos, Paula S; Brown, Elisabeth E; Kimberly, Robert P; Langefeld, Carl D (marzo de 2010). «Genetic Factors Predisposing to Systemic Lupus Erythematosus and Lupus Nephritis» [Factores genéticos que predisponen al lupus eritematoso sistémico y nefritis lúpica]. Semin Nephrol (en inglés) (Elsevier Inc) 30 (2): 164-176. PMID 20347645. doi:10.1016/j.semnephrol.2010.01.007. Consultado el 19 de febrero de 2017.
- ↑ Alarcón Riquelme ME. Genética del lupus eritematoso generalizado. ¿Qué se sabe y a dónde se va? Reumatol Clin. 2010;6:1-2. doi:10.1016/j.reuma.2009.01.002. PMID 21794669 http://www.reumatologiaclinica.org/es/genetica-del-lupus-eritematoso-generalizado-/articulo/13147008/ Archivado el 17 de septiembre de 2018 en Wayback Machine.
- ↑ a b Grygiel-Górniak, Bogna; Puszczewicz, Mariusz Jacek (septiembre de 2014). «The influence of endogenous and exogenous sex hormones on systemic lupus erythematosus in pre- and postmenopausal women» [La influencia de las hormonas sexuales endógenas y exógenas en el lupus eritematoso sistémico en la mujer pre y postmenopáusica]. Prz Menopauzalny (en inglés) (Termedia Publishing) 13 (4): 262-266. PMID 26327864. doi:10.5114/pm.2014.45003. Consultado el 19 de febrero de 2017.
- ↑ Petri, M (2008). «Sex hormones and systemic lupus erythematosus» [Hormonas sexuales y el lupus eritematoso sistémico]. Lupus (en inglés) (http://lup.sagepub.com) 17: 412-415. doi:10.1177/0961203308090026. Consultado el 19 de febrero de 2017.
- ↑ a b Maidhof, William; Hilas, Olga (abril de 2012). «Lupus: An Overview of the Disease And Management Options» [Lupus: una panorámica de la enfermedad y las opciones de tratamiento]. P T (en inglés) (Estados Unidos: MediMedia) 37 (4): 240-246, 249. PMID 22593636. Consultado el 19 de febrero de 2017.
- ↑ Barbhaiya, M; Costenbader, KH (2014). «Ultraviolet radiation and systemic lupus erythematosus» [Radiación ultravioleta y lupus eritematoso sintético]. Lupus (en inglés) (http://lup.sagepub.com) 23: 588-595. doi:10.1177/0961203314530488. Consultado el 19 de febrero de 2017.
- ↑ Perl A, Fernandez D, Telarico T, Phillips PE. Endogenous retroviral pathogenesis in lupus. Curr Opin Rheumatol. 2010;22(5):483-92. doi: 10.1097/BOR.0b013e32833c6297. PMID 20644481
- ↑ a b Lupus eritematoso inducido por medicamentos. MedlinePlus. Enciclopedia médica en español
- ↑ Joy MS, Dooley MA. Drug-induced lupus. En: Rheumatology. Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH (ed). Mosby Elsevier (2011). ISBN 978-0-323-06551-1
- ↑ Araújo-Fernández S, Ahijón-Lana M, Isenberg DA. Drug-induced lupus: Including anti-tumour necrosis factor and interferon induced. Lupus. 2014;23(6):545-53. doi: 10.1177/0961203314523871. PMID 24557776
- ↑ Aguirre Zamorano MA, López Pedrera R, Cuadrado Lozano MJ. Lupus inducido por fármacos. Med Clin (Barc) 2010:135 (3):124-129. doi:10.1016/j.medcli.2009.04.035 http://zl.elsevier.es/es/revista/medicina-clinica-2/lupus-inducido-farmacos-13151385-revision-2010 Archivado el 15 de octubre de 2014 en Wayback Machine.
- ↑ Freemer MM, King TE Jr, Criswell LA. Association of smoking with dsDNA autoantibody production in systemic lupus erythematosus. Ann Rheum Dis. 2006;65(5):581-4. PMID 16150789
- ↑ Finckh A, Cooper GS, Chibnik LB, et al. Occupational silica and solvent exposures and risk of systemic lupus erythematosus in urban women. Arthritis Rheum. 2006;54(11):3648-54.PMID 17075811
- ↑ Wang J, Kay AB, Fletcher J, Formica MK, McAlindon TE. Is lipstick associated with the development of systemic lupus erythematosus (SLE)? Clin Rheumatol. 2008;27(9):1183-7. doi: 10.1007/s10067-008-0937-6. PMID 18523821
- ↑ Enríquez-Mejía, MG (2013). «Fisiopatología del lupus eritematoso sistémico». Revista de Medicina e Investigación (en inglés) (México: www.elsevier.es) 1 (1): 8-16. Archivado desde el original el 17 de febrero de 2017. Consultado el 13 de febrero de 2017.
- ↑ Andrade F, Casciola-Rosen L, Rosen A. Apoptosis in systemic lupus erythematosus. Clinical implications. Rheum Dis Clin North Am. 2000;26(2):215-27. PMID 10768210
- ↑ a b Gatto M, Zen M, Ghirardello A, et al. Emerging and critical issues in the pathogenesis of lupus. Autoimmun Rev. 2013;12(4):523-36. doi: 10.1016/j.autrev.2012.09.003. PMID 23000207
- ↑ Crispín JC, Tsokos G. Pathogenesis of lupus. En: Rheumatology (5.ª edición). Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH (ed). Mosby Elsevier (2011). ISBN 978-0-323-06551-1
- ↑ a b c d Hahn BH, Karpouzas GA, Tsao BP. Patogenia del lupus eritematoso sistémico. En: Kelley Tratado de Reumatología (7.ª edición). Harris ED ed. Elsevier España (2006). Páginas: 1186-1212. ISBN 978-84-8174-840-6
- ↑ a b c d e f g h i j Galindo Izquierdo M. Lupus eritematosos sistémico. En: Manual SER de las enfermedades reumáticas (5.ª edición). Editorial médica Panamericana (2008). ISBN 978-849835-105-7
- ↑ a b c d e f g h Hahn BH. Lupus eritematosos sistémico. En: Harrison. Reumatología. Fauci AS, Langford CA (eds). Ed. MCGraw Hill-Interamericana (2007). ISBN 978-84-481-5576-6
- ↑ Hubscher O. Lupus neonatal. En: Lupus eritematoso sitémico. En: Lupus eritematoso sistémico (3.ª edición). Caduceo Multimedia S.L. (2009) ISBN 978-84-936320-2-1
- ↑ Reeves WH, Li Y, Zhuang H. Autoantiboidies in systemic lupus erythematosus. En: Rheumatology (5.ª edición). Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH (ed). Mosby Elsevier (2011). ISBN 978-0-323-06551-1
- ↑ Arbuckle MR, McClain MT, Rubertone MV, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349(16):1526-33. PMID 14561795
- ↑ Graham KL, Utz PJ. Sources of autoantigens in systemic lupus erythematosus. Curr Opin Rheumatol 2005;17(5):513-7. PMID 16093826
- ↑ a b c Gladman DD, Urowitz MB. Sistemic lupus erythematosus. Clinical features. En: Rheumatology. Klippel JH, Dieppe PA, (eds). Mosby-Doyma Libros S.A. (1994). ISBN 0 397 44731 0
- ↑ Pallarés Ferreres L. Esteban Marcos E, Rascón Risco J. Síntomas generales. En: Enfermedades autoinmunes sistémicas y reumatológicas. Ramos Casal M, García Carrasco M, et al. Masson S.A. (2005). ISBN 84-458-1467-2
- ↑ Gordon C. Assessing disease activity and outcome in systemic lupus erythematosus. En: Rheumatology (5.ª edición). Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH (ed). Mosby Elsevier (2011). ISBN 978-0-323-06551-1
- ↑ a b Zea Mendoza AC, Rodríguez García A, Vázquez Díaz M. Manifestaciones del aparato locomotor. En: Enfermedades autoinmunes sistémicas y reumatológicas. Ramos Casal M, García Carrasco M, et al. Masson S.A. (2005). ISBN 84-458-1467-2
- ↑ El médico interactivo. Enfermedades del tejido conectivo y vasculitis sistémicas. http://www.elmedicointeractivo.com/ap1/emiold/aula2001/tema7/vasculitis1.htm Archivado el 16 de enero de 2014 en Wayback Machine.
- ↑ a b Bielsa I, Herrero C. Manifestaciones cutáneas. En: Lupus eritematoso sistémico (3.ª edición). Caduceo Multimedia S.L. (2009). ISBN 978-84-936320-2-1
- ↑ a b c d e f g h i Schur PH, Gladman DD. Overview of the clinical manifestations of systemic lupus erythematosus in adults. UpToDate.com. 2014
- ↑ Weening JJ, D'Agati VD, Schwartz MM, et al.The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int. 2004 Feb;65(2):521-30. Review. Erratum in: Kidney Int. 2004 Mar;65(3):1132. PMID 14717922
- ↑ [1] Bomback AS, Appel GB. Diagnosis and classification of renal disease in systemci lupus erythematosus. UpToDate.com. 2014
- ↑ a b c Horga A, Sastre Garriga J, Montalbán X. Manifestaciones neuropsiquiátricas del LES. En: Lupus eritematoso sistémico (3.ª edición). Caduceo Multimedia S.L. (2009). ISBN 978-84-936320-2-1
- ↑ a b Ramachandran, Tarakad S (junio de 2016). «CNS Lupus Clinical Presentation». Medscape (en inglés). Consultado el 13 de febrero de 2017.
- ↑ Reverter Calatayud JC, Tàssies Penella D. Anticuerpos antifosfolipídicos. En: Lupus eritematoso sistémico (3.ª edición). Caduceo Multimedia S.L. (2009). ISBN 978-84-936320-2-1
- ↑ Amigo MC, Khamashta MA. Antiphospholipid (Hughes) syndrome in systemic lupus erythematosus. Rheum Dis Clin North Am. 2000;26(2):331-48. PMID 10768215
- ↑ Egües Dubuc CA, Uriarte Ecenarro M, Meneses Villalba C, et al. Hemophagocytic syndrome as the initial manifestation of systemic lupus erythematosus. Reumatol Clin. 2014;10(5):321-324. doi: 10.1016/j.reuma.2013.09.004. PMID 24316336
- ↑ Adán Civera A, Alforja Castiella MS, Molina Fernández JJ, Pelegrín Colás L. Manifestaciones oculares. En: Lupus eritematoso sistémico (3.ª edición). Caduceo Multimedia S.L. (2009). ISBN 978-84-936320-2-1
- ↑ a b c d e f g h Schur PH, Wallace DJ. Diagnosis and differential diagnosis of systemic lupus erythematosus in adults. UpToDate. 2014.
- ↑ a b Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982; 25:1271-1277
- ↑ a b Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter]. Arthritis Rheum 1997;40:1725.
- ↑ Edworthy SM. Manifestaciones clínicas del lupus eritematoso sistémico. En: Kelley Tratado de Reumatología (7.ª edición). Harris ED ed. Elsevier España (2006). Páginas: 1213-36. ISBN 978-84-8174-840-6
- ↑ Petri M. Clasificación. En: Lupus eritematoso Sistémico. 3.ª ed. (2009) Khamashta M y Vilardell M, (eds). Caduceo Multimedia. S.L. ISBN 978-84-936320-2-1
- ↑ a b Petri M, Orbai AM, Alarcón GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677-86. doi: 10.1002/art.34473. PMID 22553077
- ↑ a b c d e Úcar Angulo E, Rivera García N. Comorbilidad en lupus eritematoso sistémico. Reumatol Clin 2008;4 Supl 1:S17-21. http://www.reumatologiaclinica.org/es/comorbilidad-lupus-eritematoso-sistemico/articulo/13117215/ Archivado el 17 de septiembre de 2018 en Wayback Machine.
- ↑ Samasca G, Sur G, Lupan I, Tilinca M, Deleanu D (2014). «Celiac disease as an autoimmune condition». Cent Eur J Immunol (Revisión) 39 (3): 396-9. PMC 4440004. PMID 26155154. doi:10.5114/ceji.2014.45954.
- ↑ Denham JM, Hill ID (agosto de 2013). «Celiac disease and autoimmunity: review and controversies». Curr Allergy Asthma Rep (Revisión) 13 (4): 347-53. PMC 3725235. PMID 23681421. doi:10.1007/s11882-013-0352-1.
- ↑ Lundin KE, Wijmenga C (Sep 2015). «Coeliac disease and autoimmune disease-genetic overlap and screening». Nat Rev Gastroenterol Hepatol (Revisión) 12 (9): 507-15. PMID 26303674. doi:10.1038/nrgastro.2015.136.
- ↑ Bichile T, Petri M. Prevention and management of co-morbidities in SLE. Presse Med. 2014;43(6 Pt 2):e187-95. doi: 10.1016/j.lpm.2014.03.009. PMID 24855047
- ↑ a b c d e f g h i Schur PH, Wallace DJ. Overview of the therapy and prognosis of systemic lupus erythematosus in adults. UpToDate. 2014.
- ↑ Schoenfeld SR, Kasturi S, Costenbader KH.The epidemiology of atherosclerotic cardiovascular disease among patients with SLE: a systematic review. Semin Arthritis Rheum. 2013;43(1):77-95. doi: 10.1016/j.semarthrit.2012.12.002. PMID 23422269
- ↑ Krishnan E. Stroke subtypes among young patients with systemic lupus erythematosus. Am J Med. 2005;118(12):1415. PMID 16378793
- ↑ a b c d «Lupus. Arthritis Foundation». Archivado desde el original el 17 de junio de 2014. Consultado el 19 de septiembre de 2014.
- ↑ Ugarte, Amaia; Danza, Alvaro; Ruiz-Irastorza, Guillermo (2018-9). «Glucocorticoids and antimalarials in systemic lupus erythematosus: an update and future directions». Current Opinion in Rheumatology (en inglés estadounidense) 30 (5): 482. ISSN 1040-8711. doi:10.1097/BOR.0000000000000527. Consultado el 26 de agosto de 2019.
- ↑ a b «Consenso de la Sociedad Española de Reumatología sobre el uso de terapias biológicas en el lupus eritematoso sistémico (2013)». Archivado desde el original el 4 de marzo de 2016. Consultado el 19 de septiembre de 2014.
- ↑ Hahn BH. Tratamiento del lupus eritematoso sistémico. En: Kelley Tratado de Reumatología (7.ª edición). Harris ED ed. Elsevier España (2006). Páginas: 1237-60. ISBN 978-84-8174-840-6
- ↑ Lupus Foundation of America
- ↑ Urowitz MB, Gladman DD, Tom BD, Ibañez D, Farewell VT. Changing patterns in mortality and disease outcomes for patients with systemic lupus erythematosus. J Rheumatol. 2008;35(11):2152-8. PMID 18793004
Bibliografía
[editar]- Vasudevan AR, Ginzler EM. Clinical features of systemic lupus erythematosus. En: Rheumatology. Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH (ed). Mosby Elsevier (2011). ISBN 978-0-323-06551-1
- Hahn BH. Lupus eritematosos sistémico. En: Harrison. Reumatología. Fauci AS, Langford CA (eds). Ed. MCGraw Hill-Interamericana (2007). ISBN 978-84-481-5576-6
- Comorbilidad en lupus eritematoso sistémico. Reumatología clínica (2008)
- Consenso de la Sociedad Española de Reumatología sobre el uso de terapias biológicas en el lupus eritematoso sistémico (2013)
Enlaces externos
[editar] Wikimedia Commons alberga una categoría multimedia sobre Lupus eritematoso sistémico.
Wikimedia Commons alberga una categoría multimedia sobre Lupus eritematoso sistémico.- Lupus Foundation (enlace roto disponible en Internet Archive; véase el historial, la primera versión y la última).
- Asociación Madrileña de Lupus
- Lupus. Arthritis Foundation
- Lupus eritematoso sistémico. American College of Rheumatology
- Lupus eritematoso sistémico. MedlinePlus
- Lupus eritematoso sistémico. Ministerio de Salud de Chile
- Federación Española de Lupus
- Lupus eritematoso sistémico. Sociedad Española de Reumatología
- Lupus eritematoso sistémico. WikiSER
| Control de autoridades |
|
|---|
 Datos: Q1485
Datos: Q1485- Multimedia: Systemic lupus erythematosus / Q1485